

| << Chapter < Page | Chapter >> Page > |
Key Concepts
The original motivation for the eMalaria project was to bring together school students with university researchers in the hunt for a new anti-malarial drug. The challenge was offered, via the web, to school students to design molecules and test them using a computational drug design approach to see if the molecule might be suitable for further research as an anti-malarial drug. The participants were not merely going to be passive suppliers of computational resources in the model of the very successful cycle stealing grid drug screening systems as pioneered by Graham Richard’s group and followed most recently by the World Community Grid . Instead, they were involved with the design and selection of potential drug molecules so that they could learn more about modern approaches to drug design and development. This necessitated a significant amount of background and tutorial material to support the investigations.
At its core the eMalaria system uses the Cambridge Crystallographic Data Centre and the Gold docking engine Software . The docking program needs to investigate the whole conformational space of the potential drug and its fit into the enzyme active pocket. Gold evaluates the quality of the fit with an energy scoring function. This function takes into account the main contributions to the forces acting between the atoms in the enzyme and the potential drug; the intermolecular forces as well as any strain imposed on the drug molecule.
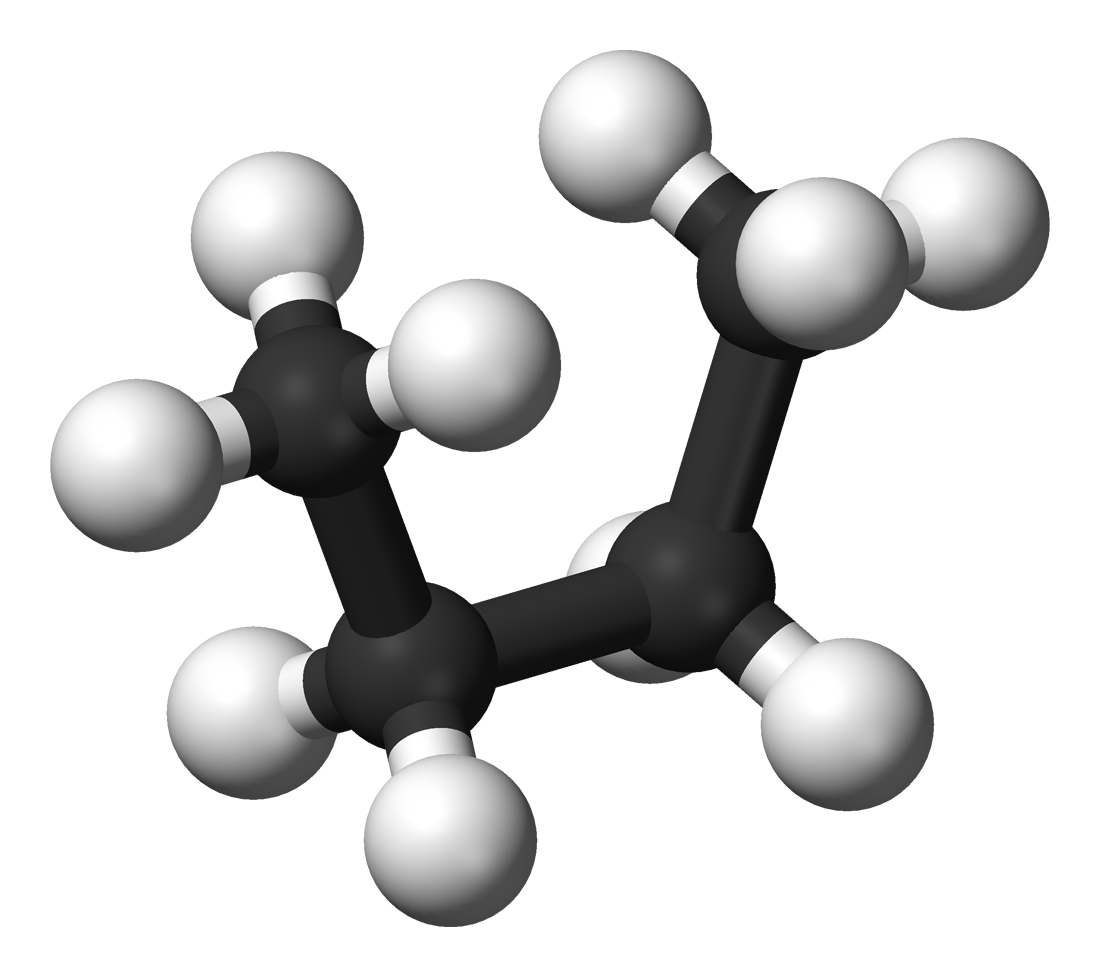
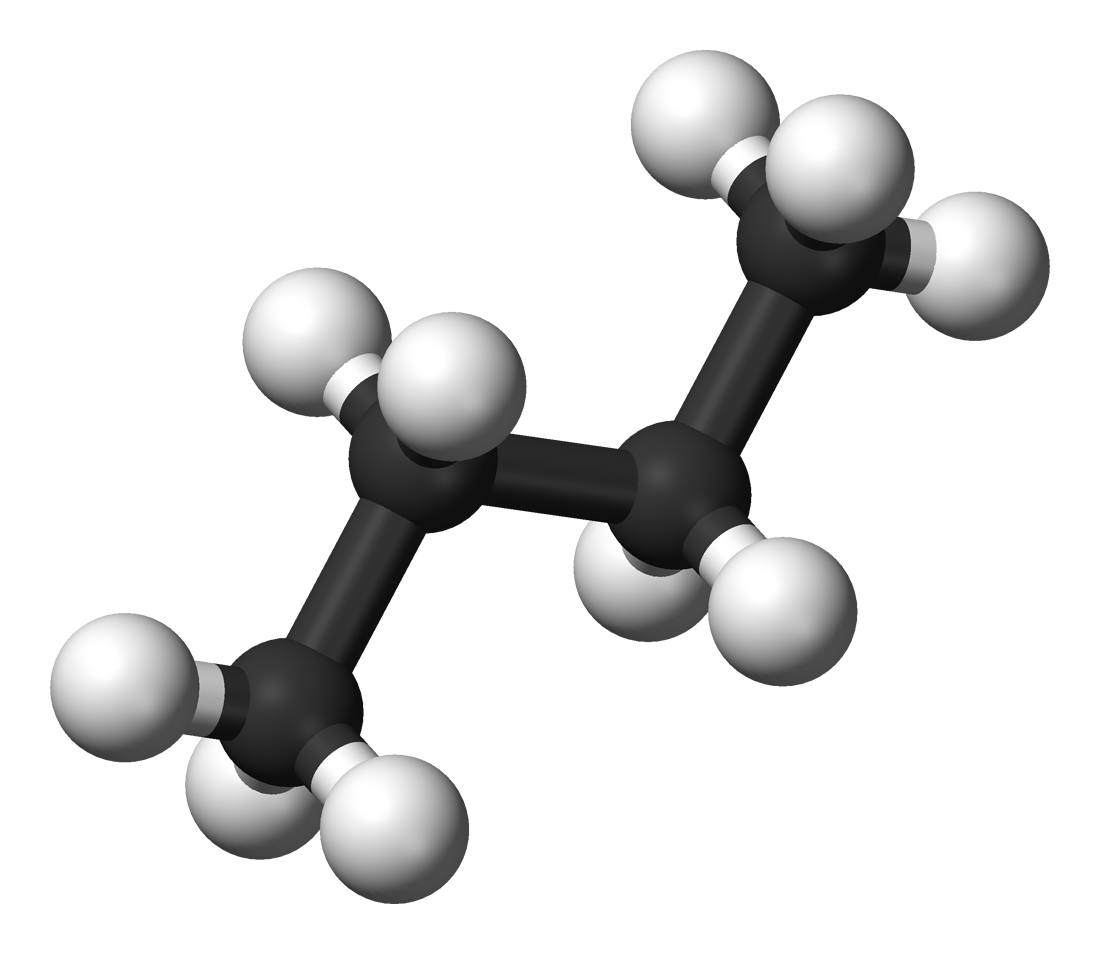
With even moderate sized molecules the size of the conformational space is very large. That is, there are very many ways in which the molecule can be twisted without breaking any bonds. Each different shape can produce different interactions between the potential drug and the enzyme pocket. It is a very significant calculation to evaluate these different possible ways in which the drug could bind into the pocket and locate the best fit.
To support the anticipated computational load and provide a model that would grow in capacity in parallel with the increase in users, we chose to use a cycle steeling grid. Several machines within the University of Southampton initially provided the computational resource. The expectation was that as schools joined the project they would provide additional computational cycles. This was made possible while protecting the commercial code by using the United Devices (UD) software, which ensured that the core Gold code was secure.
Notification Switch
Would you like to follow the 'Research in a connected world' conversation and receive update notifications?

|
|

|
|

|
|

|
|

|
|

|
|

|
|
|
|
|
|
|
|
|
|